
Cómo resolver puzles moleculares: cristalografía de rayos x
Joder, qué frío. Y otra vez lloviendo. Pero claro, es Hamburgo, estamos a principios de marzo, ¿qué esperaba? Seguro que en Madrid está haciendo un sol espléndido.
Hace ya 3 meses que dejé el laboratorio donde trabajo normalmente en Madrid para pasar una temporada en el European Molecular Biology Laboratory (EMBL) de Hamburgo. Y es que en el mundo científico es bastante frecuente cambiar temporalmente de laboratorio. La razón es simple: es imposible que todos los laboratorios tengan disponibles todas las tecnologías que existen. Algunas veces podemos llegar a ese otro laboratorio en metro pero otras, como me pasa a mí ahora, acabamos cambiándonos de país. Lo cual tampoco es una mala experiencia :smile:
¿Qué hago yo aquí entonces? Pues, en mi caso concreto, la técnica causante de que esté pasando frío (aunque disfrutando de la buena cerveza alemana) es la cristalografía de rayos X. Vale, palabro. ¿Y eso qué es? La cristalografía de rayos X es una técnica que permite determinar la estructura que forman los átomos que componen una molécula, es decir, su estructura 3D. Y esto es mucho más útil de lo que puede parecer. Yo, por ejemplo, llevo un tiempo intentando entender la función de una proteína que hay en nuestros músculos (esa es mi molécula) y conocer su estructura puede dar muchas pistas. Imaginaos por un momento que os ponen delante un objeto que no habéis visto nunca. Pues ni idea de para qué sirve. Sin embargo, si al analizar su estructura (es decir, la forma del objeto) nos damos cuenta de que tiene una pinza, probablemente tenga que ver con agarrar cosas, mientras que si tiene un gancho es muy posible que se pueda colgar. Pues algo parecido me pasa a mí cuando intento entender cómo funcionan las proteínas. Este tipo de aproximación pertenece a la rama de la biología denominada biología estructural.
Lo primero que hice cuando llegué al laboratorio de Hamburgo fue conseguir la muestra de mi proteína, lo más concentrada y lo más pura posible. Después hubo que formar cristales, que son redes tridimensionales en las que muchas copias de una molécula se organizan de forma ordenada en el espacio. Obtener cristales es fundamental para cualquier molécula que queramos analizar con esta técnica, ya sea un pequeño fármaco o un gran complejo multiproteico. Esto lo convierte también en el paso limitante, porque no todas las moléculas forman cristales fácilmente. A veces hay que esperar meses y hacer varios intentos antes de tener un cristal que podamos medir con rayos X.
Ahora la pregunta del millón. Si es un proceso tan largo y tan difícil, ¿por qué nos empeñamos en crecer cristalitos? Pues porque la interacción de los rayos X con una sola molécula produce una señal muy débil y confusa, y que no es suficiente para determinar su estructura. Sin embargo, un cristal es una estructura ordenada en la que todas las moléculas se empaquetan de manera que se forman un patrón repetitivo y en el que tienen la misma orientación en todas las direcciones del espacio. Esto hace que cuando los rayos X atraviesan el cristal en cualquier dirección, la señal que se detecta es la suma de las contribuciones de millones de moléculas. Así que en realidad los cristales tienen tanta importancia porque actúan como amplificadores de señal.
Afortunadamente mi proteína tardó muy poco en formar cristales así que no tuve que preocuparme mucho. ¡Mirad qué cosa tan bonita!
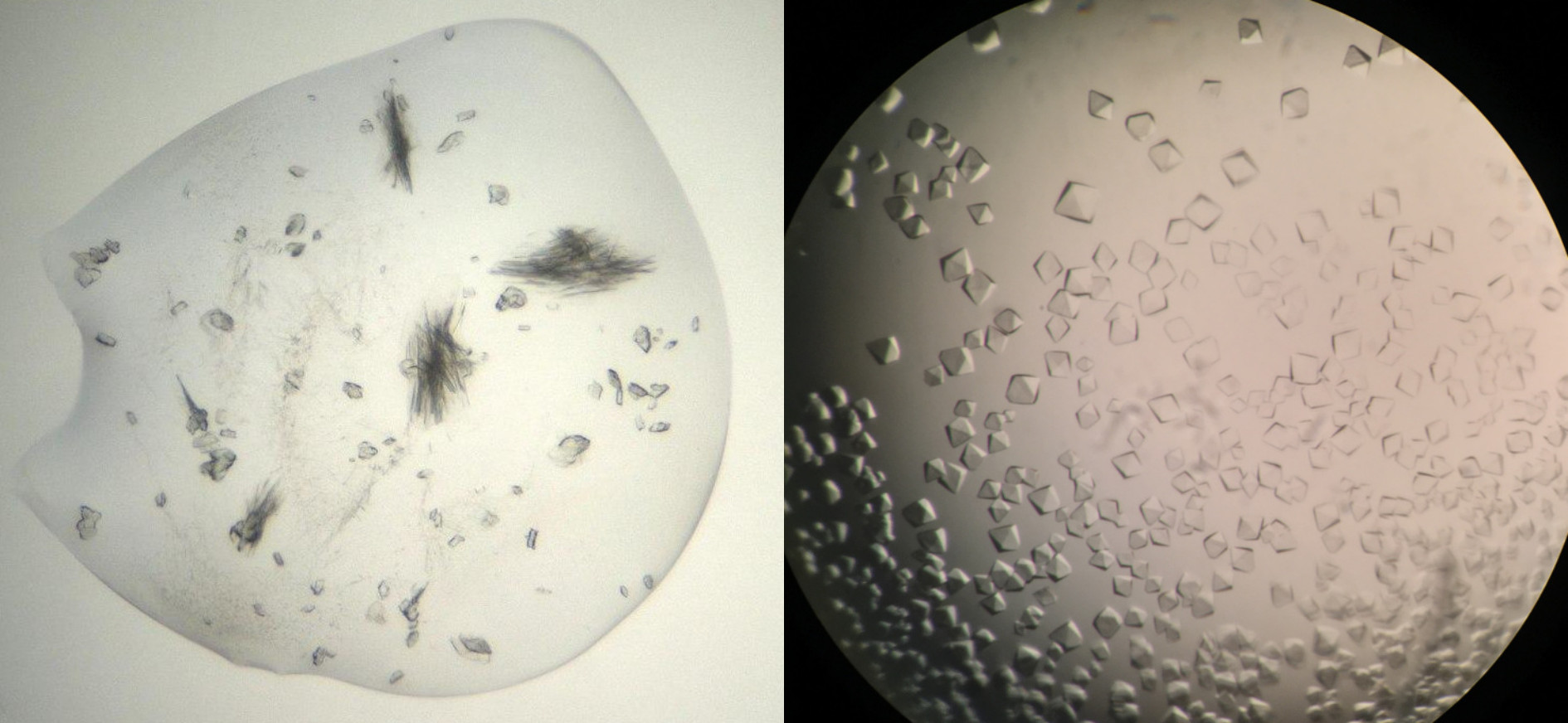
Así que ya estoy lista. A pesar del mal tiempo y de estar lejos de casa, tengo una razón para estar de buen humor. ¡Hoy toca ir al sincrotrón! Entro y todo me parece de ciencia ficción. Brazos robóticos, luces, botones, decenas de pantallas que controlan cada parámetro de lo que allí está ocurriendo… Me dan la bienvenida al Petra III, pero podrían decirme que he sido abducida y que esa es su nave espacial y me lo creería igualmente.
Una vez colocados los cristales ante la fuente de rayos X me llevan a la sala desde donde se toman las medidas. Delante de mí, una pantalla con la imagen de mi cristal esperando recibir los rayos X. A mi alrededor, más luces y botones. No sé si seré capaz de transmitiros la tensión que se siente en ese momento. El resultado de esa medida iba a determinar si mi trabajo de los últimos meses había servido para algo. Si la calidad de la muestra es buena y las moléculas están realmente ordenadas en mi cristal, obtendré el ansiado patrón de difracción con el que determinar la estructura de mi proteína. Si algo no ha salido como yo esperaba, tan solo veré un borrón y tendré que empezar de nuevo. Soy incapaz de separar mis ojos de la pantalla.
Iniciando medida… preparando fuente de radiación… irradiando muestra… cargando resultados…

¡Lo conseguí! Lo que estoy viendo en la pantalla es la estructura de mi cristal. Bueno, más o menos. La información está ahí aunque todavía no soy capaz de interpretarla. Las proteínas del cristal difractan los rayos X en un patrón de puntos característico, que es lo que se conoce como patrón de difracción. Esos puntos reciben el nombre de reflexiones, y su análisis permite determinar la distribución de los electrones en las moléculas que componen el cristal. Es decir, que sabremos que habrá átomos allá donde veamos electrones.
Para poder crear ese mapa de densidad a partir del patrón de difracción se necesitan dos tipos de información: la amplitud y la fase de los rayos X en cada reflexión. Obtener la amplitud es sencillo, se calcula a partir de las intensidades de las reflexiones medidas por el detector. Lo de las fases no es tan obvio, no se puede obtener directamente del patrón sino que hay que recurrir a métodos complementarios. En mi caso fue bastante fácil, pero como el concepto es complejo y la aproximación depende del tipo de experimento y de la molécula concreta no voy a entrar mucho en detalles. Para los curiosos dejo una serie de recursos que podéis consultar en el apartado “Para saber más”.
Una vez tenemos esos dos parámetros, amplitud y fase, podemos definir el factor de estructura, un número complejo con el que obtener el mapa de densidad electrónica en cada punto del espacio definido por las coordenadas xyz, y que se representa como ρ(xyz). La operación que relaciona la densidad electrónica con el factor de estructura se conoce como transformada de Fourier, y matemáticamente se escribe de esta manera:
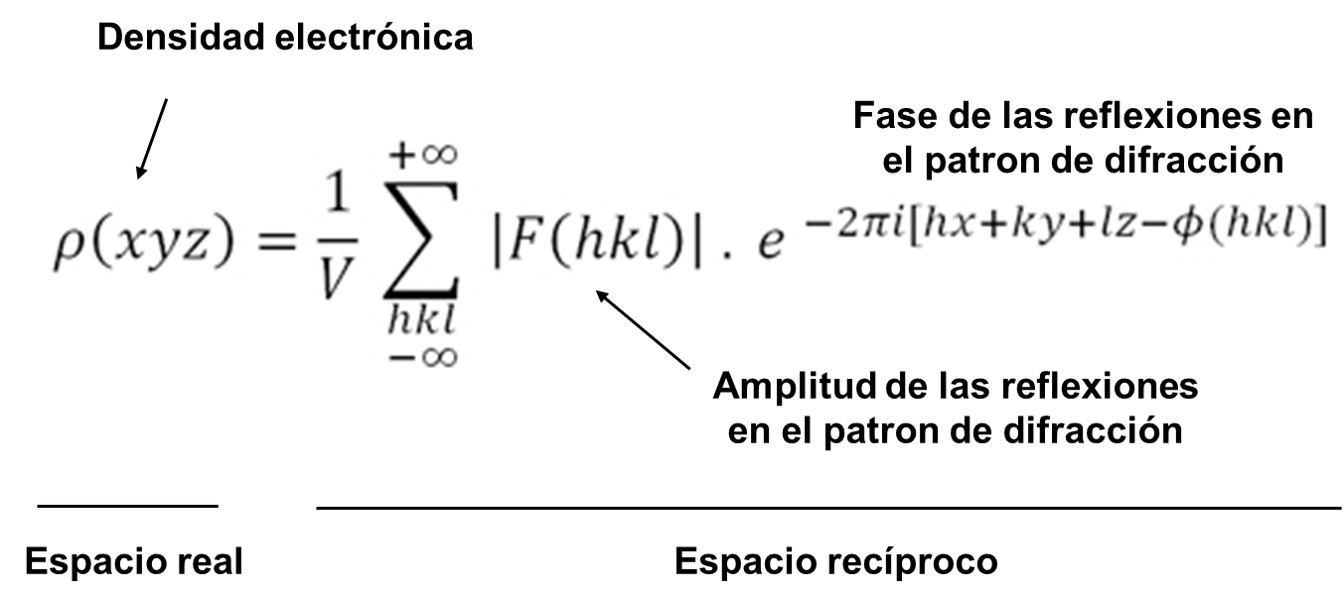
Pero ¡que no se pierda nadie! Aunque es importante entender los detalles de aquello con lo que trabajamos, el avance de la tecnología ha permitido que la mayoría de estos análisis estén bastante automatizados hoy en día. Es decir, que afortunadamente no tendré que pasarme los próximos días resolviendo esa ecuación para cada punto de mi molécula, sino que con el software adecuado podré obtenerlo de forma mucho más fácil (y por eso estaré eternamente agradecida a todas las personas y grupos de investigación que trabajan en el desarrollo de estos programas).
Lo que sí me queda son unos días de mucho ordenador y análisis. Conseguir el mapa de densidad electrónica es el primer paso, pero el trabajo no se acaba ahí. Después toca sacarle el máximo partido posible. Hasta ahora, yo sólo sabía cuáles eran las piezas que formaban mi proteína (unos bloques químicos llamados aminoácidos), pero no tenía ni idea de cómo se organizaban en el espacio. Es como si supiera cuáles son las piezas de un LEGO pero no tuviese ni idea de cómo encajan unas con otras, ni qué forma tiene la construcción final. Ahora el mapa de densidad electrónica me servirá de guía para colocar todas esas piezas y conseguir montar el LEGO.
Por ejemplo si mi pieza es así,
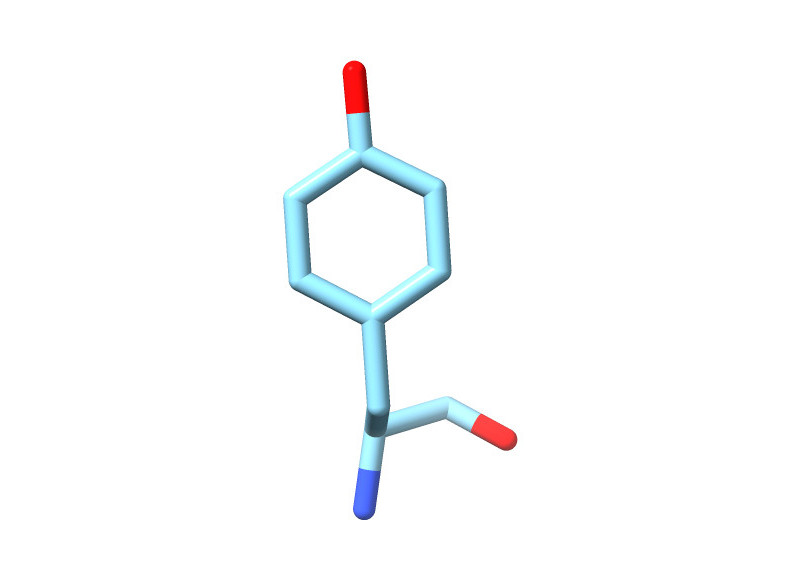
y la intento poner en esta región del mapa de densidad electrónica que he obtenido del patrón de difracción
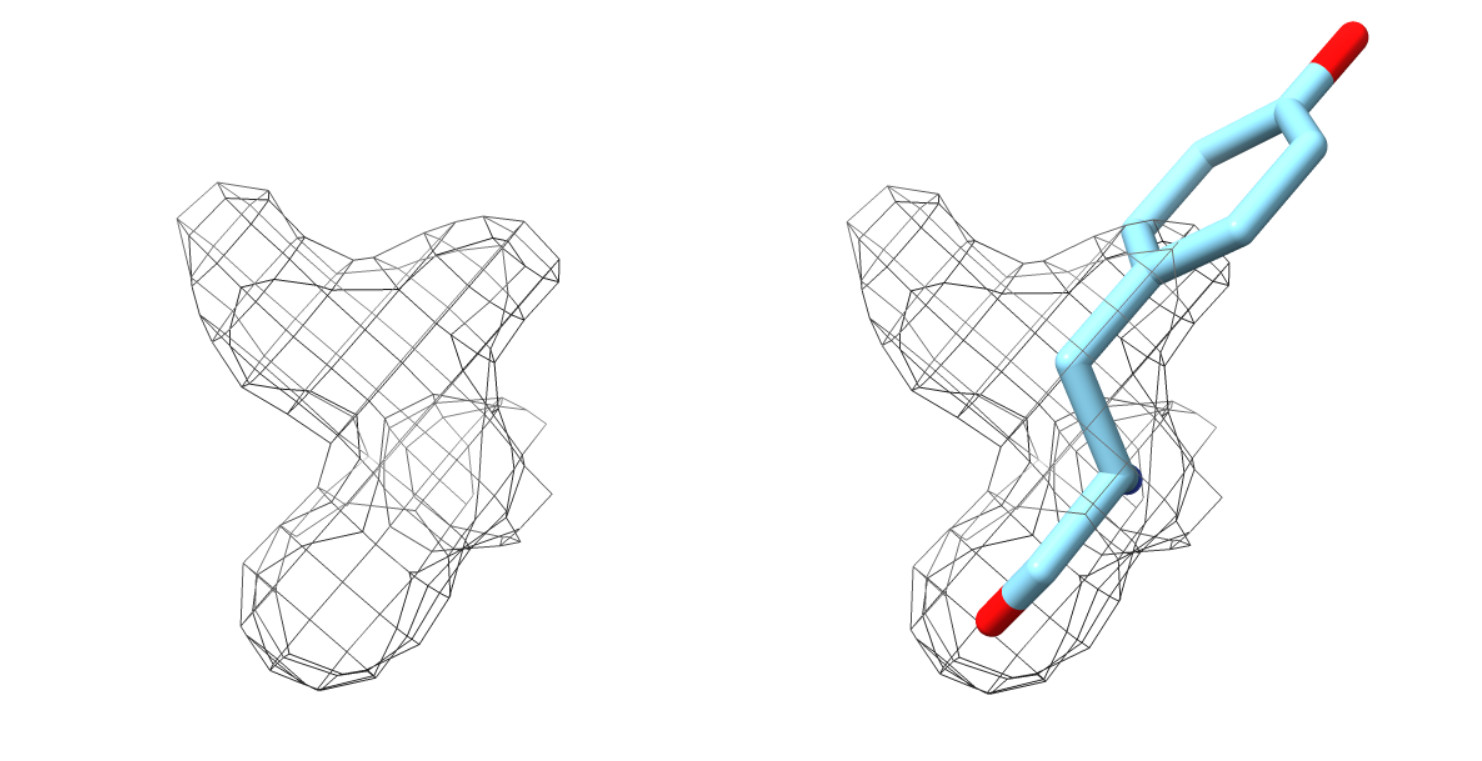
parece que no encaja. Pero si por el contrario la pongo aquí
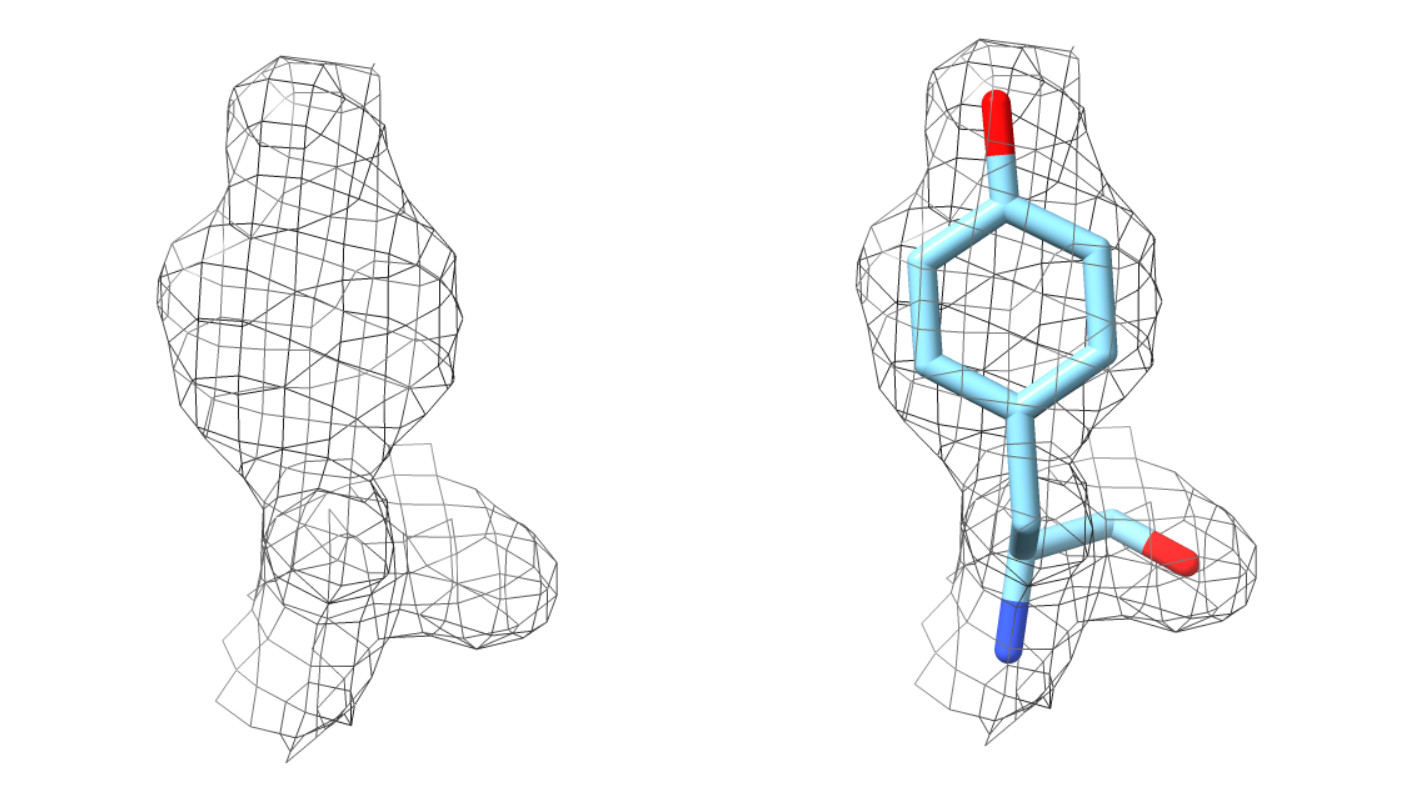
ahora ya sí encaja. Pues eso es más o menos lo que voy a estar haciendo durante los próximos días. Esperemos que mi LEGO montado pueda ayudarme un poquito a seguir averiguando cuál es la función de mi proteína ¡Deseadme suerte!
Glosario
Sincrotrón: Un sincrotrón es un tipo de acelerador de partículas, es decir, una especie de tubo muy grande en el que se utilizan campos eléctricos y magnéticos para acelerar distintos tipos de partículas (como, por ejemplo, los electrones) en una órbita cerrada. Sin embargo lo que realmente les interesa a los cristalógrafos es que las partículas cargadas, cuando se aceleran, emiten radiación, como los rayos X, que puede ser utilizada para otros experimentos. Vamos, que por muy guay y complicado que sea para un cristalógrafo es poco más que una máquina para hacer rayos X.
Petra III: El Petra III es un sincrotrón de 2.3 km de largo que se encuentra en Hamburgo (Alemania). Este acelerador es particularmente conocido por su producción de rayos X, de gran intensidad y precisión, que son utilizados por científicos de diversas áreas, desde la física a la biología molecular.
Amplitud y fase: Son parámetros que definen cómo es una onda (en nuestro caso la radiación tipo rayos X que hacemos incidir sobre el cristal de proteína). La amplitud mide el valor extremo (en positivo o negativo) que puede alcanzar esa onda. La fase nos da idea de en qué punto de la onda nos encontramos (con respecto a lo que hayamos elegido como punto de referencia). Por ejemplo, podemos tener dos ondas con la misma amplitud y frecuencia pero con distinta fase, lo que quiere decir que aunque alcancen el mismo máximo lo harán en momentos distintos.
Para saber más
- A los que os gusten las historias de rayos X y queráis saber más sobre ellos no podemos dejar de recomendaros nuestro post sobre su descubrimiento. ¡Es una historia muy curiosa!
- Si os habéis quedado con ganas de saber más sobre la cristalografía de rayos X, el departamento de cristalografía del Consejo Superior de Investigaciones Científicas tiene una página completísima para seguir aprendiendo. Aquí podéis aprender desde los fundamentos físicos de la difracción, a la química de los cristales y la construcción de modelos en biología estructural. ¡Echadle un ojo porque merece mucho la pena!
- Y si lo que queréis es una versión más sencilla que os explique la técnica en un lenguaje apto para todos los públicos podéis echarle un vistazo al post de Jerónimo Bravo en la página de la Sociedad Española de Bioquímica y Biología Molecular.
- El Protein Data Bank, una base de datos de estructuras de proteínas también tiene su propia página de divulgación, en la que explica algunos aspectos básicos (y no tan básicos) de la biología estructural: el PDB 101 (en inglés).
- Como lo prometido es deuda para los más avanzados os dejamos también un artículo de la Gaceta de la Real Sociedad Matemática Española, (ISSN 1138-8927, Vol. 17, Nº 4, 2014, págs. 705-716) sobre el problema de las fases.
- Los cristales de proteína pueden tener formas preciosas. Tanto que, ¡existe un premio para el cristal más bonito del año! Os dejamos el link al ganador del premio de 2019.¿No os parece un cristal precioso?
- Pero no sólo los cristales son bonitos. Las estructuras de los complejos proteicos que se encuentran en nuestras células tienen formas de lo más variopinto. El centro de regulación genómica de Barcelona tiene un póster en el que podéis ver algunas de estas máquinas moleculares.